In cancer single agent drug treatment is rarely, if ever, curative due to drug resistance. We are developing new technologies and approaches to think about drug resistance and generate new drug combinations for use in cancers.
In one approach (Donnella et al) we mapped kinase activation following
drug exposure to understand how pathways are rewired by drug treatment and identify barriers to efficacy of anti-cancer drugs. This approach revealed an exciting new combinatorial approach to treat breast cancers using the combination of Aurora kinase and PI3K inhibitors. In the future
we anticipate that modeling pathway dynamics and drug adaptation will be critical for the design of effective new combination therapies. In a second study we identified Aurora Kinase A as a critical driver is tumor adaptation, residual disease and acquired resistance in non-small cell lung cancer (NSCLC) (Shah et al.). Aurora kinase inhibitors and EGFR inhibitors are synergystic by exploiting a fascinating reduncancy in the control of apoptotic machinery that tumor cells exploit to escape single agent drug treatment. Together, these findings reveal the previously unappreciated and criticalrole of Aurora Kinase A on resistance to targeted therapy. Clinical studies testing drug combinations are planned.
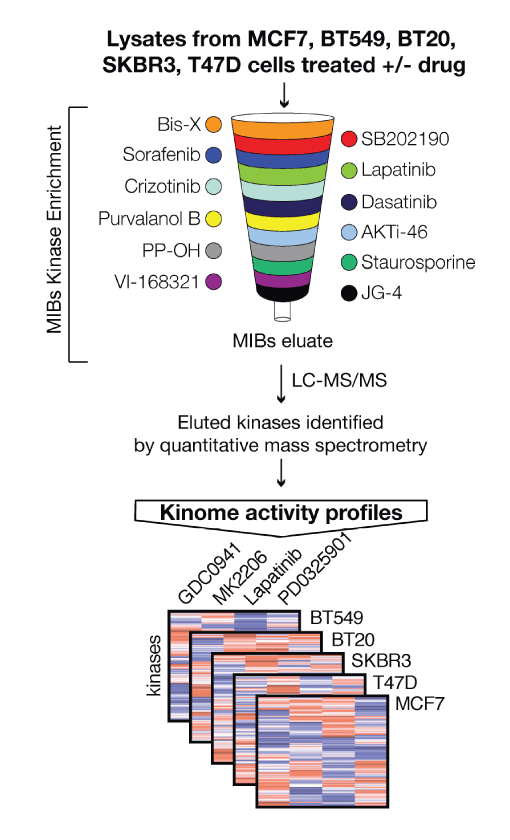

A major barrier to our ability to make more effective treatments is our incomplete knowledge of cancer pathways and the contexts in which they are active. We develop approaches
to generate new protein-protein interaction data using affinity purification followed by mass spectrometry as well as computationally analyze interactome data and integrate it with cancer
patient data.
We developed a computational algorithm to integrate multi-platform -Omics data from breast cancer to identify 219 gene modules that capture the majority of molecular variation in this disease. We use this approach
to delineate molecular signals coming from the tumor microenvironment and those that are preserved in cancer cell lines. These networks predict cancer drug responses and can serve as robust, high resolution biomarkers for therapies.

An approach to identify network modules in cancer and use them as biomarkers for therapeutics using Modular Analysis of Genomic Modules in Cancer (MAGNETIC). From
Webber et al.